Common Cold Coronaviruses
Most people are now familiar with the cold-causing coronaviruses, so in this post, I'm going to discuss some information related to their origins and evolution.
Human Coronaviruses
There are 4 cold-causing coronaviruses (CoVs) that are endemic (actively circulating) in humans, and together, they cause ~15% of common cold cases. They are named human coronavirus (HCoV-) OC43, NL63, 229E, and HKU1, and a significant fraction of the human population has antibodies to multiple coronaviruses, suggesting a global distribution 1.
The first human coronaviruses were discovered in 1965 from people suffering from colds not caused by previously identified agents, such as influenza, parainfluenza, adenoviruses, respiratory syncytial virus, enteroviruses, and rhinoviruses 2. These coronaviruses were clustered in the group that came to be known as 229E. Subsequently, these 229E samples from sick individuals were given to healthy volunteers and found to cause colds in the recipients 3. Even though they only caused mild illness in the donor patients, this would NOT have been an ethical experiment by today’s standards.
OC43 was discovered in the 1960s as well, and until the emergence of SARS-CoV in 2002, it was believed that all coronaviruses capable of infecting humans caused only mild disease 4. NL63 was first isolated from a child in 2004 in the Netherlands 5, and HKU1 was discovered in Hong Kong in 2005 from an elderly patient with pneumonia returning from Shenzhen, China 6.
The four “mild” human coronaviruses can cause severe disease in the very young, the very old, and immunosuppressed individuals 7. Many people develop severe lower respiratory tract infections, particularly in the winter months when CoV incidence peaks, and coronaviruses are among the causes of pneumonia and hospitalization in infants 8,9. As animals age, the alveoli (grape-like sacs where gas exchange occurs) in the lungs become less elastic, respiratory muscles weaken, and the cough reflex is diminished. All of these could make it easier for older adults to become infected with all respiratory pathogens, not just coronaviruses. Infants do not have fully developed immune systems yet, making them susceptible to severe disease.
Phylogenetic Placement
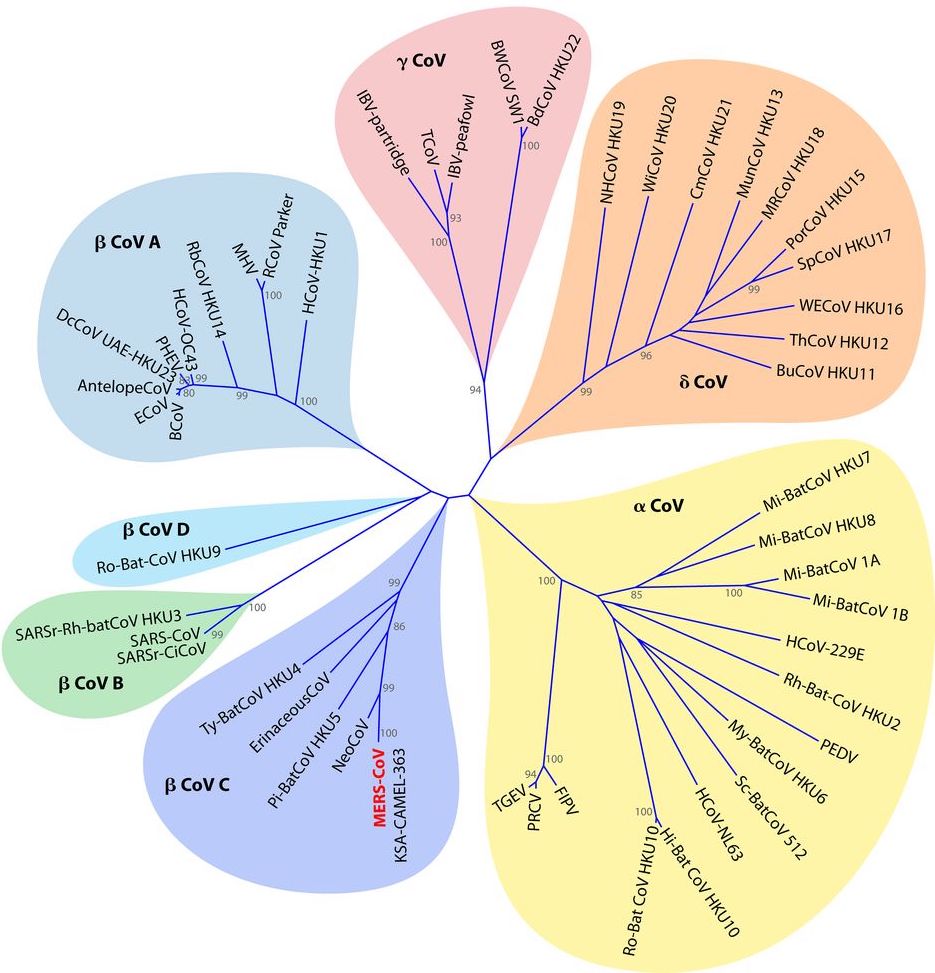
Coronaviruses fall into one of four genera (plural of genus): alpha (α), beta (β), gamma (γ), and delta (δ). NL63 and 229E are α-coronaviruses, and OC43, HKU1, SARS-CoV, SARS-CoV-2, and MERS-CoV are all β-coronaviruses. Alpha- and beta-CoVs are likely to have originated in mammals, and gamma- and delta-CoVs in birds, though the viruses now infect all types of vertebrates. Gamma- and delta-coronaviruses have been primarily found in birds, fish, and marine mammals, and the similarities between viruses found in different species suggest past or even ongoing cross-species transmissions 10.
The β-coronavirus genus has four subgenera -- A, B, C, and D. In 2018, they were renamed as Embecovirus (lineage A), Sarbecovirus (lineage B), Merbecovirus (lineage C) and Nobecovirus (lineage D), and a 5th subgenus, Hibecovirus, was added 11. The following table shows common CoVs in each of the 4 main β-coronavirus subgenera:
| β-CoV A (Embecovirus) | β-CoV B (Sarbecovirus) | β-CoV C (Merbecovirus) | β-CoV D (Nobecovirus) |
|---|---|---|---|
| Mouse hepatitis virus | SARS-CoV | MERS-CoV | HKU9 |
| OC43 | SARS-CoV-2 | HKU4 | |
| HKU1 | SARS-related CoVs | HKU5 |
The phylogenetic tree below was constructed using the sequences of the RNA-dependent RNA polymerase from several coronaviruses. Analysis of different viral proteins will yield slightly different results in how closely related certain CoVs are to each other, but general groupings and relative positions are approximately the same at the genome level.
Gastrointestinal Involvement
Coronaviruses infect a wide variety of animals, and in addition to respiratory disease, they cause enteric (intestines), hepatic (liver), and neurologic symptoms. For example, Swine Acute Diarrhea Syndrome coronavirus (SADS-CoV), Porcine Enteric Diarrhea Virus, and Transmissible Gastroenteritis coronavirus cause primarily intestinal symptoms in pigs and are extremely lethal for piglets. Coronaviruses that cause intestinal symptoms in animals are generally severe in young animals because their gastrointestinal tracts are not fully developed and able to protect against infection. The stomach pH tends to be less acidic, and digestive enzymes are less abundant, so the environment is less hostile to pathogens. These enteric-specific coronaviruses can not infect or do not transmit well between humans, but the respiratory human coronaviruses can also cause symptoms of gastrointestinal (GI) disease, including in many COVID-19 patients.
The receptors of coronaviruses are widely expressed on enterocytes (intestinal cells), and many can infect and replicate in the GI tract. Indeed, in human organoids -- small models of human organs studied in the lab -- both SARS-CoV and SARS-CoV-2 replicated inside enterocytes and induced production of cytokines and interferon-stimulated genes 12. MERS-CoV was also found to replicate in intestinal epithelial cells and organoids, and direct infection of mice via the gut resulted in lethal MERS-CoV infection 13.
Although coronaviruses have been found to replicate in the gut, it is unclear why the vast majority of human deaths are due to respiratory failure. Some hypotheses are 1) differences in mucosal immunity between the gut and respiratory epithelia and 2) the shorter lifetime of intestinal cells than respiratory cells that may lead infected gut cells to be naturally removed by the host more quickly.
Origins of Human Coronaviruses
Alpha- and beta-coronaviruses are the only genera that have been isolated from humans. While both are predominantly found in bats, a subset of beta-CoVs may have a rodent origin. Like beta-CoVs, alpha-Covs have a bat origin and can also infect humans, rodents, pigs, dogs, and cats. With the exception of SARS-CoV and SARS-CoV-2, human coronaviruses are not closely related to each other within the Coronaviridae family. They originated in other animals (typically bats or rodents) and were eventually able to infect humans and become permanent viruses in the population through separate transmission events. Homology between sequences from different animals can point to their origins. For example, human HKU1 is most closely related to mouse hepatitis virus, suggesting a rodent origin of this CoV.
Using the sequence difference between two viruses and the known replication rates of CoVs based on the accuracy of their polymerases (RNA-copying enzymes), scientists can calculate approximately how long ago two virus sequences shared a common ancestor. When the viruses were separated into different host species, they diverged, or underwent separate evolutionary trajectories. This back-calculating analysis is called molecular clock analysis.
SARS-Like CoVs
Many SARS related coronaviruses (SARSr-CoVs) are found in Rhinolophus (horseshoe) bat populations in southern China. A paper from 2017 found that the genetic pieces of the SARS-CoV genome could all be found in virus isolates from horseshoe bats in a single cave in Yunnan, China 14. The authors speculate that recombination (or mixing) of these isolates in bats or an intermediate host could have given rise to SARS-CoV; therefore, no single SARSr-CoV was the culprit. In addition, many isolates had different spike proteins that all bound to human ACE2, meaning that they could emerge into humans again, just like SARS-CoV-2.
Alpha CoVs: NL63 and 229E
A virus from a Hipposideros bat in Ghana and human CoV 229E are predicted to have shared a common ancestor between 1680 and 1800, suggesting an African origin of 229E 15. However, in 2007, acute respiratory disease and sudden death were documented in captive alpacas in California 16, and an alphacoronavirus genetically closely related to 229E was later identified as the cause 17. This alpaca CoV was most similar to 229E samples isolated from the 1960s-1980s, so the authors hypothesized that transmission between humans and alpacas may have occurred during that time, but which animal transmitted it to the other is unknown.
Genes of the alpaca CoV clustered either with bat viruses or between bat viruses and 229E 18. This has led to speculation that camelids are the intermediate host of 229E precursors. However, this is unproven because 229E-like viruses have been found in bats in Ghana, while alpacas are native to South America. Further complicating matters is the fact that alpaca coronavirus was isolated from captive alpacas in California, and we do not know if this coronavirus is found in wild alpaca populations. Other camelids may offer some clues, so analyses of 229E-like viruses in dromedary camels, which are found in Africa and the Arabian peninsula, have been undertaken. Samples from camels in the Arabian Peninsula (Saudia Arabia and the United Arab Emirates) had more antibodies for 229E, alpha-CoVs, and distant 229E-related bat CoVs than samples from camels in Africa (Kenya, Somalia, Sudan, and Egypt) 19. The camel-derived viruses used the same receptor as 229E to enter cells, and 229E antibodies neutralized the camel viruses and prevented infection of human cells.
Based on genetic analyses, the authors of the study concluded that HCoV-229E could have originated from a mixed pool of camelid viruses because 229E is most similar to one branch. Both MERS-CoV and 229E-related viruses cause mild or no disease in camels, which could suggest that these viruses have infected camels for a long time. Generally speaking, the longer a host and virus coexist, the less pathogenic the virus is because the host develops immunity. A virus may mutate to be less severe if this simultaneously increases its transmisibility, which is the strongest selective pressure. Then the alpacas of California, which sickened and died from a related coronavirus, were likely exposed more recently.
The origins of NL63 have been more difficult to pin down. The closest scientists have come is isolating similar viruses in bats, the putative animal of origin, but we don’t know when and where cross-species transmissions may have occurred. Viruses similar to NL63 have been found in African bats of the Hipposideros genus, and NL63 in humans is likely the result of recombination between NL63- and 229E-related viruses 20.
Recently, alpha-coronaviruses have been isolated from bats in North America (no beta-CoVs though) 21. These CoVs were detected in fecal samples from bats in the Rocky Mountain region, but are not very closely related to other alpha-CoVs found in Asian bats. Another study found alpha-CoVs in the intestines and lungs of hibernating North American little brown bats, which did not show virus-induced inflammation 22. These results suggest that the bats are persistently infected with the viruses, but without symptoms, similar to Asian bats infected with SARS-like viruses. NL63 has also been shown to infect and replicate in cells from the North American tricolored bat, so human-to-bat transmission could occur, or a reverse zoonosis 23.
Mild Beta CoVs: OC43 and HKU1
The mostly closely related CoV to OC43 is the bovine (cow) coronavirus. Molecular analyses indicate that these two coronaviruses shared a common ancestor at the end of the 19th century, when the ancestor of both spilled from cows into humans 24. OC43 is also related to mouse hepatitis viruses, and since the bovine coronavirus is found between human and murine CoVs in sequence relatedness, scientists hypothesize that the ancestor was transmitted from rodents to cows and then to humans.
Some coronaviruses more similar to bovine CoVs than OC43 have been isolated from humans. These patients typically suffered from enteric disease without respiratory symptoms, another telltale sign of animal CoVs 25. It is interesting to see viruses jump the species barrier in real time, but also a warning of the inevitability of future outbreaks of zoonotic diseases.
Since the closest viral relatives to HKU1 are mouse hepatitis virus and rat coronavirus, scientists speculate that it originated in rodents. A study that isolated a coronavirus from the rat species used in laboratories, Rattus norvegicus, suggested that this isolate may point to rodent ancestors of all β-CoVs of lineage A 26.
MERS-CoV
The emergence of MERS-CoV is not well understood. In 2012, the virus was isolated from a man in Saudi Arabia who died of pneumonia and kidney failure 27. Subsequent analyses found that it was genetically most similar to HKU4 and HKU5, coronaviruses found in bats in Hong Kong 28. Partial genome sequences from viruses in Ghana, Mexico, and Europe show a high degree of similarity to MERS-CoV, but without complete genome sequences, the true level of similarity is difficult to tell 29,30.
HKU4 and HKU5 were isolated from bats native to East Asia, but not to the Arabian peninsula. However, antibodies to MERS-CoV were found in camels as early as 1983, so the virus was circulating in camels before it either acquired the ability to infect humans or conditions were right for it to cross species in 2012 31. MERS-like CoVs likely have a long history with Arabian camels, as evidenced by the mild disease they cause in these animals. These CoVs may have been transmitted from bats to camels long ago when the ranges of the these animals were significantly different from what they are today. Greater surveillance of bats and camelids in Europe and Asia is required to determine the origins of MERS-CoV.
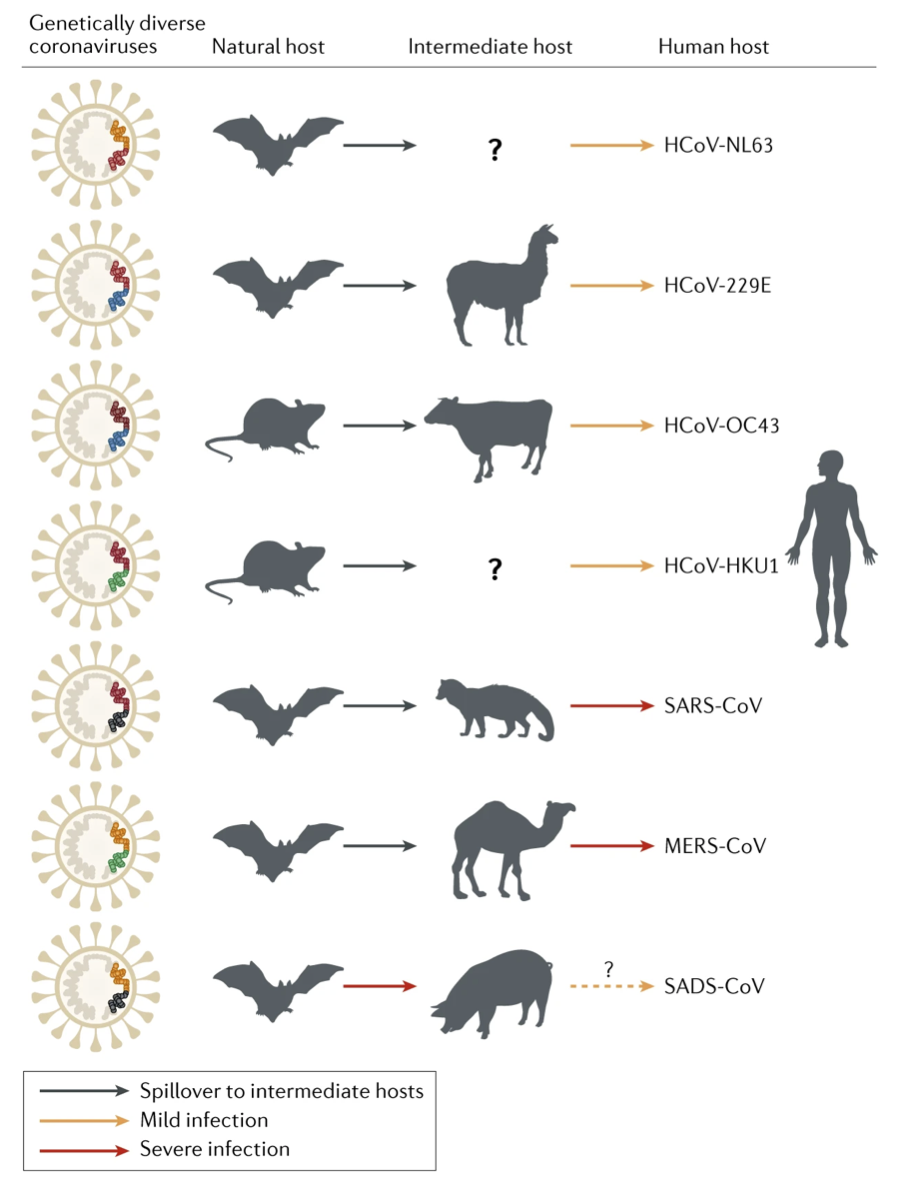
Source: Figure 2, Cui et al., 2019. This figure is a summary of the zoonotic origins of human coronaviruses and one recently identified swine coronavirus, SADS-CoV. Since the paper was published in early 2019, SARS-CoV-2 is not included.
One caveat to note in molecular clock studies is that there is usually a reference sequence that is taken to be the “prototype” of a particular virus. Virus isolates vary considerably because as they replicate in individual hosts, they acquire mutations, so for the purposes of determining evolutionary relatedness between viruses, standardization is required across scientists. However, viruses like 229E and OC43 were isolated in the 1960s and passaged (grown) through many cell lines before they were eventually sequenced decades later using advanced molecular biology techniques. During the many cycles of growth, they would have gained more mutations, possibly skewing molecular clock analyses.
Seasonality of CoVs
The four mild respiratory infection-causing human coronaviruses show seasonal patterns like influenza. In the Northern hemisphere, their incidence peaks in the winter months (January - March), which may be attributed to increased viral stability, human behavioral changes that keep them inside more often, and impaired immune activation at colder temperatures 32. Many American politicians were hopeful that warmer months would dampen the COVID-19 pandemic, but seasonality is only a factor for pathogens that are ACTIVELY circulating in humans, not new pathogens to which the population does not have immunity.
Here, we must distinguish between prevalence and incidence. Prevalence is the TOTAL number of people with a certain disease at a given time, while incidence is the number of NEW cases in a time interval. HIV, hepatitis viruses, and tuberculosis have high prevalence, along with noncommunicable diseases like diabetes, cardiovascular disease, and cancer. All of these diseases are frequently of long duration (months to years), so the number of new cases is greater than the number of people who are removed from the total disease pool by recovery or death. Pathogens with very high incidence are typically those that cause explosive outbreaks, such as Ebola, Nipah, and severe coronaviruses. These diseases spread rapidly and cause dramatic increases in infection, but resolve relatively quickly in individual patients (resolve could be recover or die), so the overall prevalence of the infection does not grow.

Many diseases have both high prevalence and incidence. There were approximately 38 million people worldwide with HIV/AIDS in 2019 (high prevalence), and an estimated 1.7 million individuals worldwide became newly infected with HIV that same year (high incidence) 33. The good news is that HIV incidence is decreasing; the 2019 incidence represents a 23% decline in new HIV infections since 2010. Cancer has both high prevalence and incidence, but the incidence is increasing. There were 17-18 million new cancer diagnoses worldwide in 2018 34,35, and this is projected to increase to more than 23 million diagnoses per year in 2040 36. The number of people living within 5 years of a cancer diagnosis in 2018 was more than 43 million.
Seasonality is only significant for infectious diseases with low to medium incidence in the human population. Seasonality can change the transmissibility and stability of the virus and how well our immune systems respond. But for a novel virus like SARS-CoV-2, an immunologically naive population more than compensates for any reductions of transmission in warmer weather. Pandemic pathogens (high incidence) benefit from hosts that have no pre-existing immunity, so transmissibility and lethality will be high. On the other hand, viruses like influenza that have been circulating in humans for centuries will wax and wane with the seasons because the host population isn’t as susceptible to infection as it is to SARS-CoV-2 infection. Influenza is more appropriately called seasonal influenza because pandemic influenza, to which the population does not have any pre-existing immunity, will not follow seasonal patterns because of a highly susceptible population.
Conclusion
It is likely that SARS-CoV-2 will join the mild coronaviruses as an actively circulating coronavirus. This doesn’t mean that the virus itself becomes weaker or less lethal, but the human population will build up immunity. We know from COVID-19 and many other viral illnesses that children fare better on average than adults. Children are still infectious, spread a lot of virus particles, and can develop fatal syndromes, but the younger immune systems of children generally mount an innate immune response faster to control viral replication early on. Children also build up their immune systems by exposures to pathogens. They get sicker often because they have fewer circulating memory T and B lymphocytes that protect them from future infections, but they have more naive lymphocytes ready to respond to new threats.
SARS-CoV-2 is here to stay, but as it circulates in the population, more children will be exposed and have some degree of immunity in the future. This may not be sterilizing immunity, in which an infection is prevented altogether, but will likely lead to milder symptoms. The safest way to do this is through a vaccine, rather than through careless, or even deliberate, exposure.
When novel pathogens with high transmissibility and low-to-medium lethality emerge into hosts, widespread epidemics ensue because the hosts are immunologically naive, and the virus can spread silently to a large fraction of the population. After sufficient spreading, the pathogen becomes another seasonal strain owing to viral mutations and host immunity. For example, one of the actively circulating influenza A strains, H1N1, is derived from the 2009 pandemic strain.
Immunity to the mild coronaviruses wanes in 1-2 years, so people get future mild infections, which is likely what we will see with SARS-CoV-2. Influenza vaccines are given every year to account for changes in the virus genome, but SARS-CoV-2 does not mutate or recombine as much as influenza does. However, it will take years for SARS-CoV-2 to become another mild virus, so to reduce the percentage of infected people who develop severe or fatal cases, we need vaccines. The vaccines may not prevent infections, but have a very good chance of reducing the severity of symptoms. Hopefully, broadly protective vaccines and the exposures that have occurred already will prevent severe cases of reinfection. Only time will tell though.
Leave a Comment